


Pour les identifier, le consortium COVID-19 Host Genetics Initiative a analysé les génomes de près de 50 000 patients, et les a comparés avec ceux de 2 millions de contrôles non infectés. Gilles Darcis est infectiologue et chercheur au FNRS (fonds de la recherche scientifique) au sein du département des sciences cliniques de la faculté de médecine de l’Université de Liège. Il nous présente cette étude sans précédent, à laquelle il a participé aux côtés de 3500 autres chercheurs et médecins.
The Conversation France : Le consortium COVID-19 Host Genetics Initiative a réalisé une méta-analyse basée sur les données de 49 562 patients COVID-19 et 2 millions de contrôles. D’où provenaient ces données ?
Gilles Darcis : Ces travaux, qui ont été coordonnés par Andrea Ganna, chef de groupe à l’Institut de médecine moléculaire de Finlande de l’Université d’Helsinki, et Mark Daly, directeur du FIMM et membre de l’Institut Broad du MIT et de Harvard, ont utilisé des données fournies par les centres de recherche participant au consortium. Celles-ci provenaient d’études menées dans 19 pays.
Tous les centres qui ont participé ont créé une « biobanque » à partir d’échantillons provenant de patients infectés par la Covid-19. Concrètement, tout patient infecté et hospitalisé, que ce soit en salle normale ou en salle de soins intensifs, se voyait proposer de participer à ce projet. Concrètement, il s’agissait d’accepter qu’un échantillon de sang soit prélevé puis stocké (sous forme de plasma, de cellules sanguines ou de sérum), de manière à pouvoir procéder à différentes analyses, à des fins de recherche.
Si le patient acceptait, l’ADN de ses échantillons était séquencé et analysé. Ses données cliniques étaient également associées (avait-il été hospitalisé en soins intensifs, placé sous oxygène, intubé, combien de temps, etc.), le tout étant bien entendu anonymisé. Dans notre centre, la participation a été très forte, les refus ont été exceptionnellement rares. Certains patients ont même ressenti une certaine fierté à l’idée de pouvoir apporter leur aide à la recherche.
De cette façon, les chercheurs ont pu corréler, grâce à des analyses statistiques, les particularités des génomes des participants avec le fait d’avoir développé une forme de Covid plus ou moins sévère (trois catégories ont été retenues : les formes critiques définies comme celles qui ont nécessité une assistance respiratoire à l’hôpital ou ont entraîné le décès du patient ; les formes modérées ou sévères, qui concernaient les hospitalisations « simples » ; les formes sans sévérité, où l’infection par le coronavirus SARS-CoV-2 avait simplement été constatée après test).
Le travail de séquençage a été colossal, puisque l’on parle ici de plusieurs dizaines de milliers de patients : les données de 61 biobanques provenant de 19 pays ont été utilisées.
Ces données ont été comparées avec celles de patients qui n’avaient pas infectés par le Covid. Ces données « contrôles » provenaient d’autres biobanques, créées pour des recherches concernant d’autres maladies (au moment où les patients acceptent de céder leurs échantillons à des fins de recherche, il leur est demandé s’ils acceptent qu’ils puissent être utilisés pour d’autres projets de recherche – même plusieurs années plus tard – ou s’ils ne veulent participer qu’au projet dans le cadre duquel est effectué le prélèvement).
TC : Qu’ont révélé ces analyses, et qu’est-ce qui fait la force de cette étude ?
GD : 13 emplacements intéressants ont été mis en évidence dans le génome des patients (ndlr : on parle de loci – pluriel de locus : un locus est un emplacement unique sur le chromosome définissant la position d’un gène ou d’une séquence d’ADN. À un même locus, la séquence d’ADN peut varier d’un individu à l’autre).
Ces 13 régions du génome sont en effet pour la plupart associées à une sensibilité accrue à l’infection par le coronavirus SARS-CoV-2, certaines étant également associées avec un risque accru de développer une forme sévère de la maladie. Une minorité d’entre elles était même associée uniquement à un risque de développer un Covid sévère (sans augmentation de la sensibilité au SARS-CoV-2).
Ces résultats ont pu être obtenus car cette étude a porté sur des nombres de patients beaucoup plus importants les études précédentes : les plus importantes n’incluaient « que » quelques milliers de patients, ce qui est déjà beaucoup, tandis que celle-ci en a impliqué des dizaines de milliers.
La mise en évidence de ces associations dépend évidemment de la taille de l’échantillon. Étant donné qu’il contient énormément de participants qui ont le Covid, il est parfois plus facile statistiquement de mettre en évidence une association avec le risque d’attraper le Covid que de développer une forme grave, en se focalisant uniquement sur les patients qui étaient hospitalisés pour cette raison en soins intensifs.
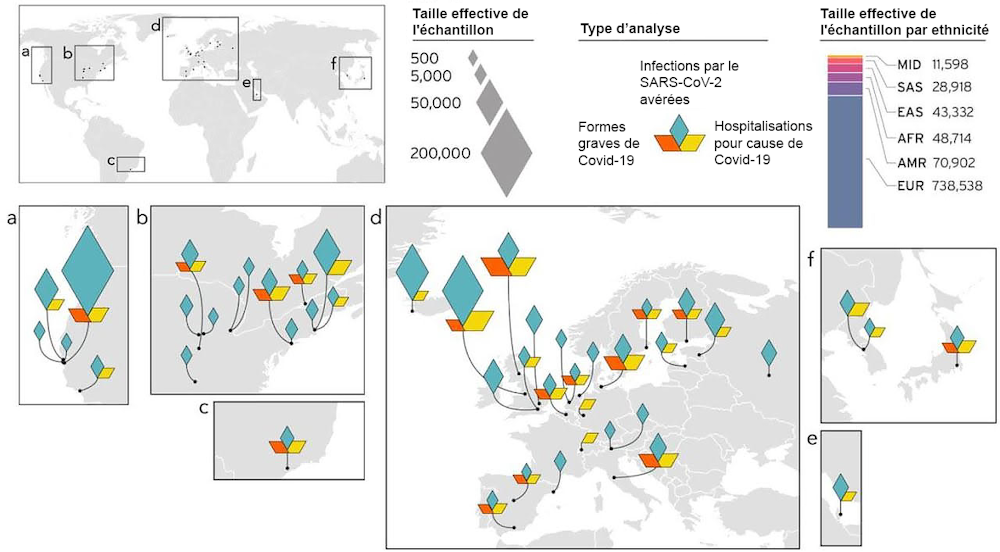
L’autre grande force de ces travaux est la diversité des profils génétiques qu’elle a inclus. Les études précédentes étaient généralement basées sur des données provenant de centres de recherches situés au sein d’un même pays, ou dans deux ou trois pays différents. La diversité génétique était alors limitée. Ici, l’étude a eu lieu sur plusieurs continents : elle a impliqué des centres de recherche situés en Amérique du Nord, en Amérique du Sud, en Europe et en Asie.
Cette diversité a permis d’identifier certains loci qui n’auraient pas été remarqués dans d’autres conditions. C’est par exemple le cas d’un locus situé à proximité du gène FOXP4 (connu pour son implication dans le cancer du poumon). Ce locus a été associé à la sévérité de la Covid-19 chez 32 % des patients originaires de populations d’Asie du Sud-Est, alors que ce n’est le cas que chez 2 à 3 % des patients d’origine européenne. Sans l’inclusion de ces populations de patients asiatiques, ce locus serait passé inaperçu.
TC : Quelles sont les implications cliniques de cette étude ?
GD : Certains de ces 13 loci sont associés à d’autres maladies des poumons, comme la fibrose pulmonaire ou le cancer du poumon, ainsi qu’à des maladies auto- immunes et inflammatoires. Cela confirme que les facteurs influant sur l’activation du système immunitaire et l’inflammation sont très importants dans la Covid-19, très probablement parce qu’ils concourent aux lésions pulmonaires.
Ces travaux ont aussi permis d’affiner certaines connaissances : ainsi, la région 3p21.31 du génome, qui avait été associée par d’autres travaux avec le risque de développement de formes sévères, était considérée comme un seul locus. Cette étude a montré qu’en réalité, deux loci très proches l’un de l’autre existaient à cet endroit. Autrement dit, deux cibles thérapeutiques potentielles.
Toutefois, pour l’instant, ces résultats n’ont pas d’application clinique directe. En revanche, ils donnent des pistes importantes pour orienter les recherches ultérieures vers certaines voies métaboliques plutôt que d’autres. L’idée est de pouvoir par la suite identifier de potentielles cibles thérapeutiques en lien avec l’inflammation par exemple, ou la capacité du virus d’infecter les cellules.
On peut ensuite imaginer mettre au point de nouveaux médicaments visant ces cibles, ou tester sur elles des médicaments existants, pour les repositionner dans la lutte contre la Covid-19 s’il s’avéraient avoir un effet.
TC : Pourrait-on imaginer appliquer cette approche à d’autres maladies infectieuses ?
GD : Tout à fait. De précédentes études ont montré qu’il existe des prédispositions d’origine génétique en matière de risques de développer des formes sévères de grippe ou d’hépatites virales. Ces résultats ont été obtenus grâce à des études similaires, mais souvent moins robustes, car basées sur des échantillons de moindre taille. Cette nouvelle approche pourrait permettre de les approfondir.
Toutefois, monter un projet d’une telle ampleur est un véritable défi, non seulement technique (traiter les échantillons sanguins requiert par exemple d’avoir accès à des laboratoires spécifiques, où ils peuvent être manipulés en toute sécurité), mais aussi en matière de collecte et d’harmonisation des données cliniques, notamment. Un patient classé sévère ou modéré dans un centre ne le sera pas forcément dans un autre, les critères pouvant différer. En ce qui concerne les données biologiques, les techniques de mesure ne sont pas non plus tout à fait les mêmes partout. Les techniques de séquençage peuvent aussi varier d’un laboratoire à l’autre. L’harmonisation est donc essentielle, pour limiter la variabilité – et donc les biais – dans la collecte et l’analyse des données.
Mais il faut souligner un point important : au-delà de leur intérêt scientifique, ces travaux ont été rendus possibles grâce à une collaboration internationale sans précédent, qui a permis d’obtenir des résultats en un temps record. Par ailleurs, vous noterez que la publication scientifique qui en a résulté n’est signée que du consortium « The COVID-19 Host Genetics Initiative » : aucun auteur ou laboratoire n’est particulièrement mis en avant. Dans un milieu aussi compétitif que celui de la recherche scientifique, c’est très rare.
Quand on voit la quantité d’articles scientifiques sur la pandémie qui ont été publiés rapidement, sans revue par les pairs, pour être rétractés ensuite, on se dit que la compétition n’est pas toujours souhaitable… Cette aventure scientifique hors-norme est probablement aussi une des raisons qui ont suscité l’intérêt d’une revue scientifique aussi prestigieuse que Nature.
Si l’on était capable de fournir les mêmes efforts pour d’autres pathologies, qu’elles soient d’origine infectieuse ou non, il ne fait aucun doute que les connaissances progresseraient rapidement.




{kind=link}